
EncoMPASS
Encyclopedia of Membrane Proteins Analyzed by Structure and Symmetry
 Enter the 4-character identifier from the Protein Databank for your structure of interest (or XXXX_A for chain A of that structure) or use the Advanced Search to search by name or other features. Enter the 4-character identifier from the Protein Databank for your structure of interest (or XXXX_A for chain A of that structure) or use the Advanced Search to search by name or other features.
|
Advanced Search |
Updated: August 13, 2025 9:00 |
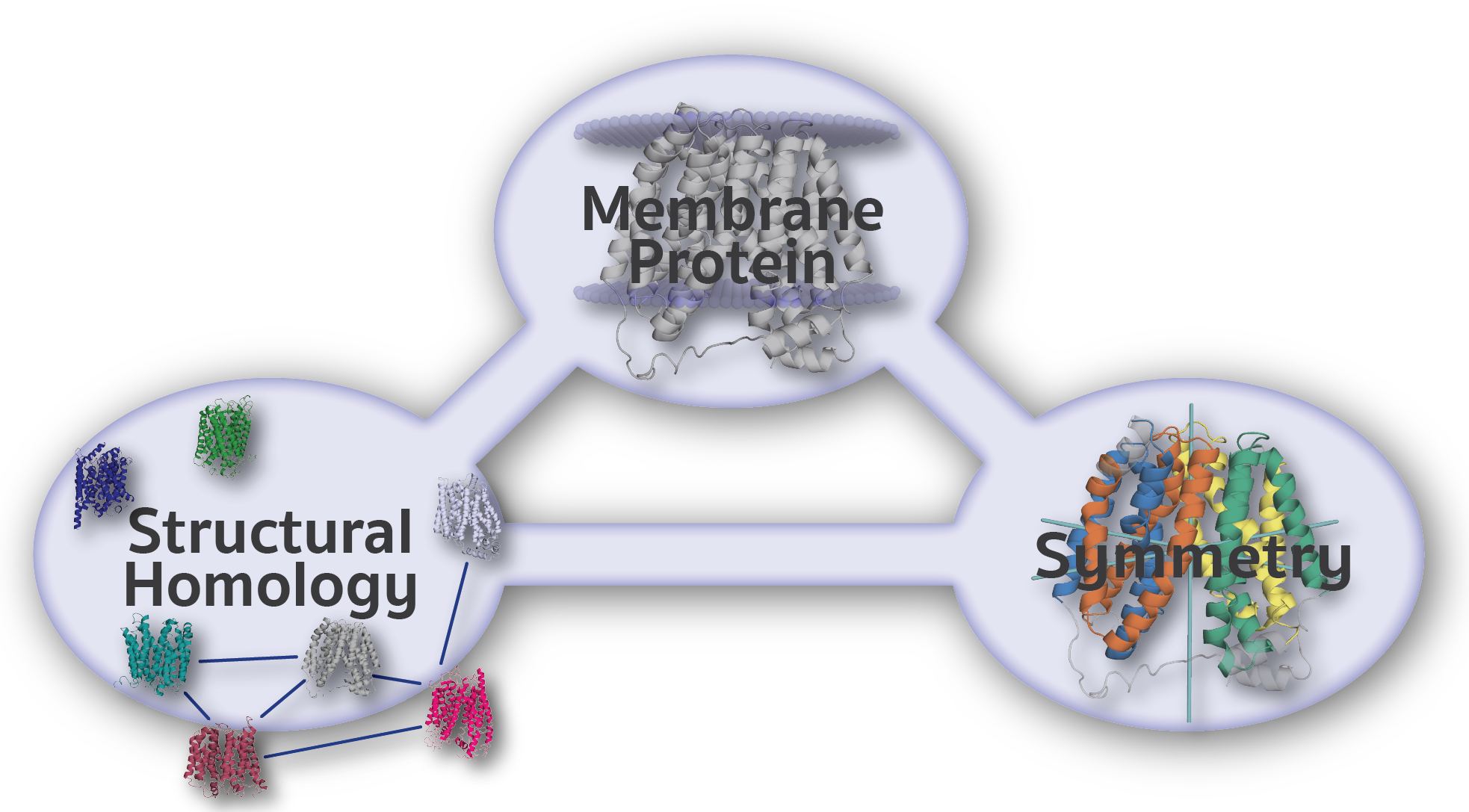
The Encyclopedia of Membrane Proteins Analyzed by Structure and Symmetry (EncoMPASS) is an automated database for relating integral membrane proteins of known structure from the points of view of their sequence, structure, and quaternary and internal symmetries. Analysis is presented for all membrane-spanning PDB entries, and separately for all membrane-spanning chains found therein.
NIH | NINDS | USA.Gov | Disclaimer
NINDS Copyright 2017